Isao Masuda, Simon D. P. Baugh, Kevin McClay, Ryan J. Ford, Lily Tayag, Eric D. Strobel, Thomas Grant, Samantha A. DeSando, Hongjuan Guo, Thomas Christian, Diana Bao Hang, Zheng Ruan, John L. Kulp III, Katie B. Freeman, Ya-Ming Hou, Allen B. Reitz, and Dipti Kanabar.
ACS Medicinal Chemistry Letters. (2026) June 10. DOI: doi.org/10.1021/acsmedchemlett.6c00164.
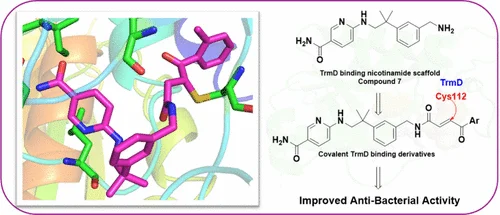
Bacterial tRNA (m1G37) methyltransferase (TrmD) is an essential enzyme required for accurate translation and represents an attractive antibacterial target distinct from its eukaryotic counterpart, Trm5. Here, we describe the development of nicotinamide-based TrmD inhibitors incorporating electrophilic Michael-acceptor motifs to enhance cellular antibacterial activity. Mass spectrometry-based peptide mapping shows covalent modification of TrmD at Cys112, a residue proximal to the AdoMet (S-adenosylmethionine) binding region, providing a mechanistic basis for covalent target engagement. Consistent with this mechanism, electrophile-containing analogues showed improved antibacterial activity compared with the parent scaffold. In bacterial growth inhibition assays, selected compounds displayed greater functional specificity for TrmD-driven growth suppression relative to human Trm5, supporting preferential bacterial target engagement. Collectively, these results establish Cys112 as a chemically addressable site for covalent inhibition of TrmD and provide a foundation for the development of TrmD-directed antibacterial agents.
Zheng Ruan, Junuk Lee, Yangyang Li, Ian J. Orozco, Juan Du & Wei Lü.
Nat Chem Biol. (2026) Jan 5. DOI: doi.org/10.1038/s41589-025-02097-7.
TRPM5 is a Ca2+-activated monovalent cation channel essential for taste perception, insulin secretion and gastrointestinal chemosensation. Canonical TRPM5 activation requires Ca2+ binding at two distinct sites: an agonist site within the lower vestibule of the S1–S4 pocket in the transmembrane domain (CaTMD) and a modulatory site in the intracellular domain (CaICD) that tunes voltage dependence and agonist sensitivity. Here we characterize CBTA as a noncalcium agonist that binds to the upper vestibule of the S1–S4 pocket, directly above CaTMD. CBTA alone mimics the dual role of CaTMD and CaICD, merging agonist activation with voltage modulation. CBTA also renders TRPM5 supersensitive to Ca2+, synergistically hyperactivating the channel even at near-resting Ca2+ levels. We further demonstrate that the inhibitor triphenylphosphine oxide binds the same site but stabilizes a nonconductive state. These opposing effects reveal the upper S1–S4 pocket as a multifunctional regulatory hub integrating activation, inhibition and modulation in TRPM5.
The cryo-EM density maps of CBTA/EDTA–TRPM5(consensus), CBTA/EDTA–TRPM5(open), CBTA/Ca2+–TRPM5, 200 μM Ca2+–TRPM5, 1 μM Ca2+–TRPM5, 2 μM Ca2+–TRPM5, CBTA/EGTA–TRPM5, Ca2+–TRPM5-D797A, CBTA/Ca2+–TRPM5(D797A), CBTA/Ca2+–TRPM5-D797N, TPPO/Ca2+–TRPM5, CBTA/Ca2+–TRPM5-E337A, CBTA/Ca2+–TRPM5-Q771A and Ca2+–TRPM5-Q771A were deposited to the EM Data Bank under accession numbers EMD-71244, EMD-71245, EMD-71246, EMD-71247, EMD-71248, EMD-71249, EMD-71250, EMD-71251, EMD-71252, EMD-71253, EMD-71254, EMD-71255, EMD-71256 and EMD-71257, respectively.
The structure models of EDTA/CBTA–TRPM5(open), Ca2+/CBTA–TRPM5, EGTA/CBTA–TRPM5, Ca2+–TRPM5-D797A, Ca2+/CBTA–TRPM5-D797A, Ca2+/CBTA–TRPM5-D797N, Ca2+/TPPO–TRPM5, Ca2+/CBTA–TRPM5-E337A, Ca2+/CBTA–TRPM5-Q771A and Ca2+–TRPM5-Q771A were deposited to the PDB under accession codes 9P3N, 9P3O, 9P3P, 9P3Q, 9P3R, 9P3S, 9P3T, 9P3U, 9P3V and 9P3W, respectively.

Yangyang Li*, Zheng Ruan*, Junuk Lee, Ian J. Orozco, Edward Zhou, Juan Du & Wei Lü.
Nat Commun. (2025) Dec 11;16(1):11057. DOI: 10.1038/s41467-025-66028-9.
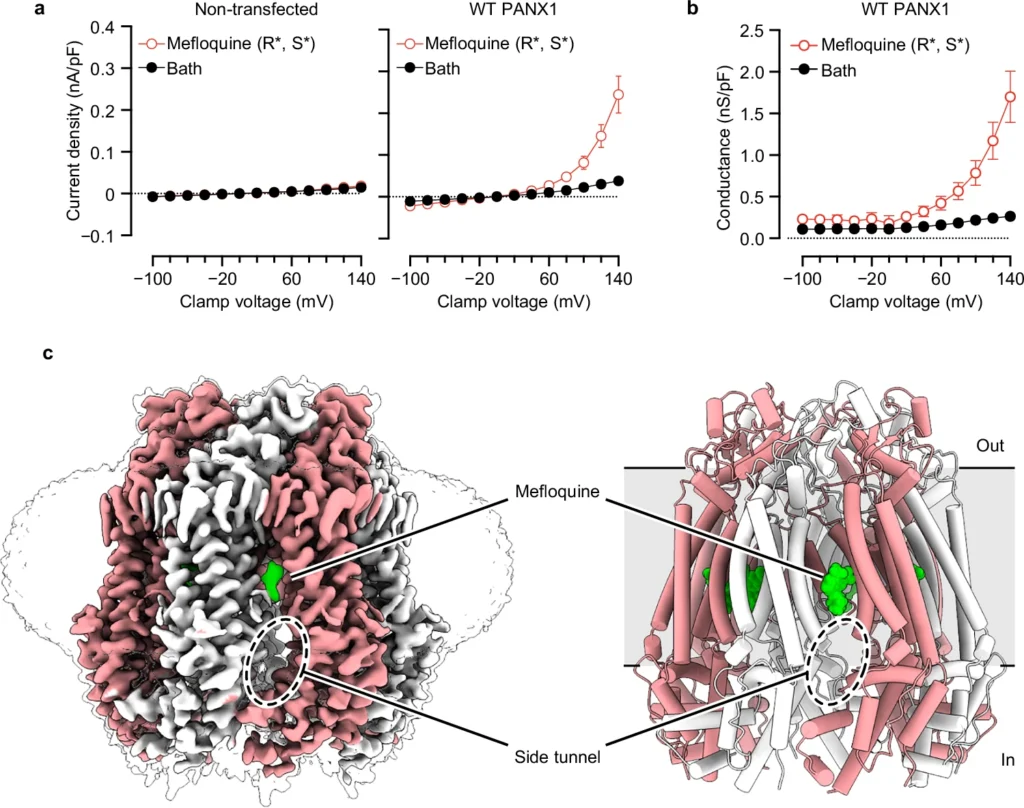
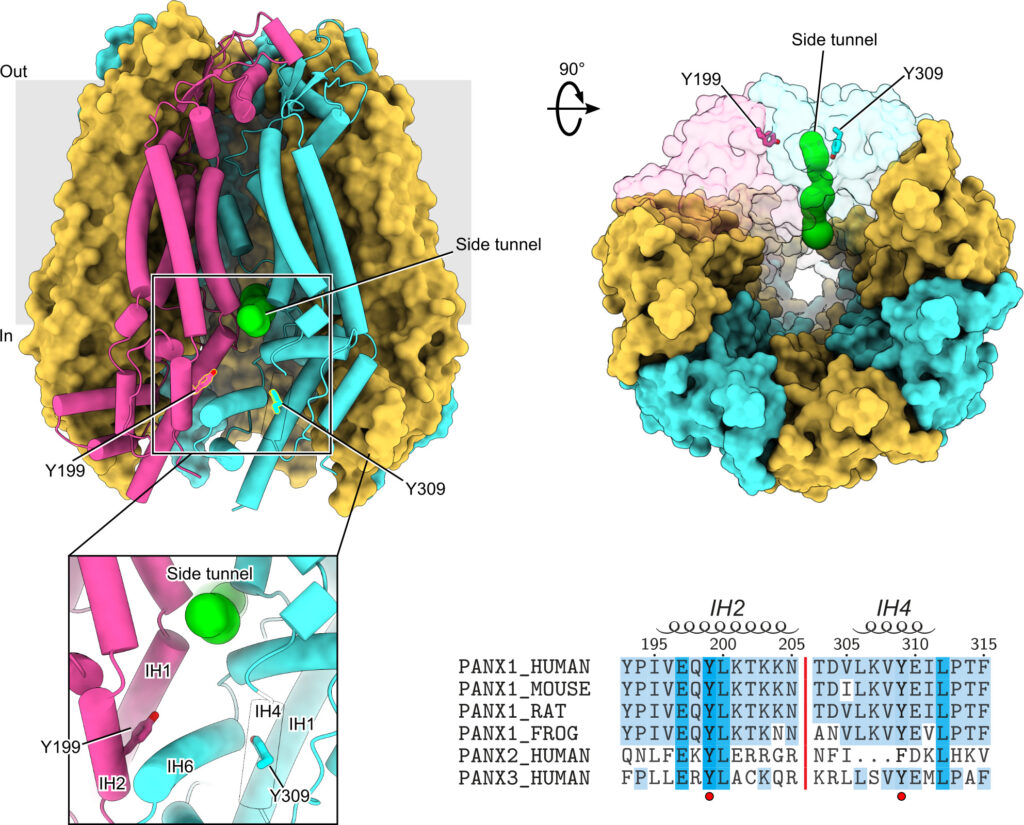
Purinergic signaling relies on ATP release through exocytosis and large-pore channels. Large-pore channels permeate both small anions like chloride and large signaling molecules like ATP, but how this broad cargo selectivity is structurally controlled remains elusive. Here we investigate PANX1, a prototypical large-pore channel, and uncover structural plasticity at the extracellular entrance formed by seven tryptophan (W74) residues. The W74 sidechains are flexible, sampling conformations that range from a constricted state permissive only to chloride to a dilated state compatible with ATP. These states are coupled to variable cation–π interactions between W74 and arginine 75 (R75), suggesting a mechanism for dynamic tuning of pore architecture and selective cargo permeation. We also identify mefloquine as a positive modulator of PANX1 that binds near the side tunnel to control ion flow through this pathway. Together, these findings define the structural principles underlying PANX1 permeation and modulation.
EMD-70760 (PANX1 (0 mM ATP) consensus)
EMD-70761 (PANX1 (10 mM ATP) consensus)
EMD-70762 (PANX1 (20 mM ATP) consensus)
EMD-70763 (PANX1 (30 mM ATP) consensus)
EMD-70764 (PANX1 (merged 0, 10, 20, 30 mM ATP datasets) consensus)
EMD-70765 (PANX1 (putative ATP-bound))
EMD-70766 (PANX1 (Apo))
EMD-70767 (PANX1 (Constricted pore))
EMD-70768 (PANX1 (Dilated pore))
EMD-70769 (PANX1 (1 mM mefloquine))
EMD-70770 (PANX1–W74A (10 mM ATP))
9OQG (PANX1 (0 mM ATP) consensus)
9OQH (PANX1 (10 mM ATP) consensus)
9OQI (PANX1 (20 mM ATP) consensus)
9OQJ (PANX1 (30 mM ATP) consensus)
9OQK (PANX1 (merged 0, 10, 20, 30 mM ATP datasets) consensus)
9OQL (PANX1 (putative ATP-bound))
9OQM (PANX1 (Apo))
9OQN (PANX1 (Constricted pore))
9OQO (PANX1 (Dilated pore))
9OQP (PANX1 (1 mM mefloquine))
9OQQ (PANX1–W74A (10 mM ATP))
Rahil Taujale, Sung Jin Park, Nathan Gravel, Saber Soleymani, Rayna Carter, Kennady Boyd, Sarah Keuning, Zheng Ruan, Wei Lü, Natarajan Kannan.
eLife. (2025) Nov 18: 14:RP106134. DOI: doi.org/10.7554/eLife.106134.2.
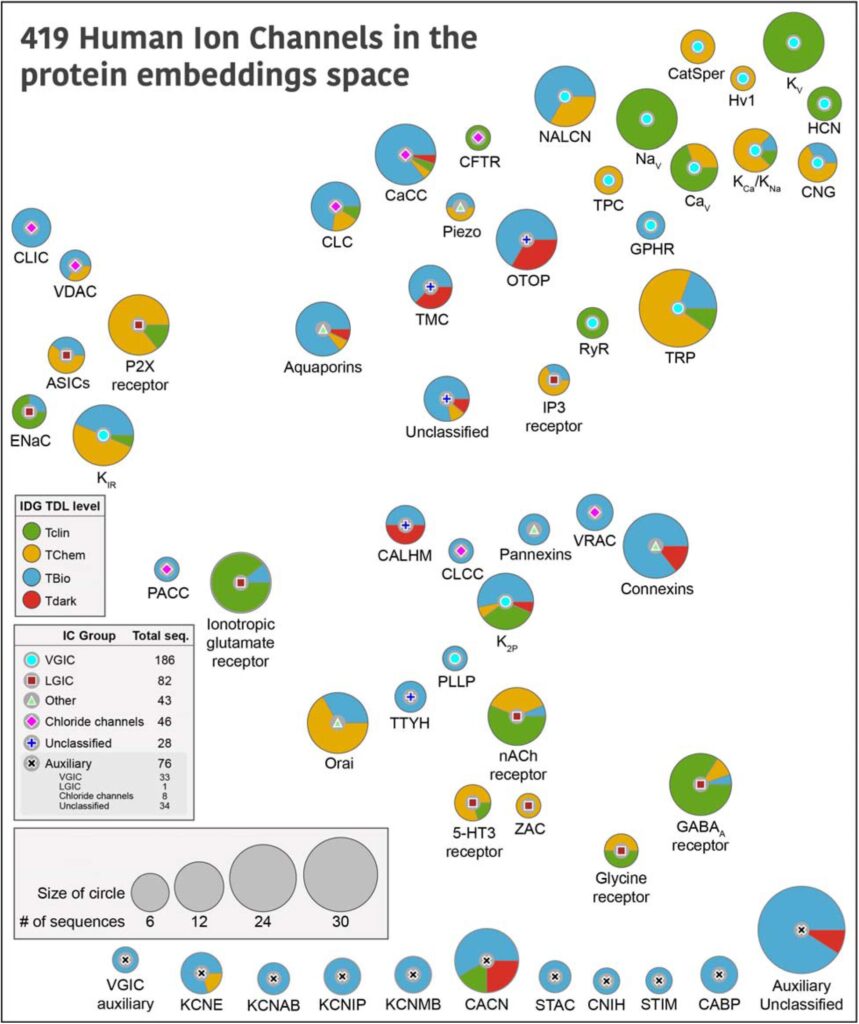
The ion channel (IC) genes encoded in the human genome play fundamental roles in cellular functions and disease and are one of the largest classes of druggable proteins. However, limited knowledge of the diverse molecular and cellular functions carried out by ICs presents a major bottleneck in developing selective chemical probes for modulating their functions in disease states. The wealth of sequence data available on ICs from diverse organisms provides a valuable source of untapped information for illuminating the unique modes of channel regulation and functional specialization. However, the extensive diversification of IC sequences and the lack of a unified resource present a challenge in effectively using existing data for IC research. Here, we perform integrative mining of available sequence, structure, and functional data on 419 human ICs across disparate sources, including extensive literature mining by leveraging advances in large language models to annotate and curate the full complement of the “channelome”. We employ a well-established orthology inference approach to identify and extend the IC orthologs across diverse organisms to above 48,000. We show that the depth of conservation and taxonomic representation of IC sequences can further be translated to functional similarities by clustering them into functionally relevant groups, which can be used for downstream functional prediction on understudied members. We demonstrate this by delineating co-conserved patterns characteristic of the understudied family of the Calcium Homeostasis Modulator (CALHM) family of ICs. Through mutational analysis of co-conserved residues altered in human diseases and electrophysiological studies, we show that these evolutionarily-constrained residues play an important role in channel gating functions. Thus, by providing new tools and resources for performing large comparative analyses on ICs, this study addresses the unique needs of the IC community and provides the groundwork for accelerating the functional characterization of dark channels for therapeutic intervention.
Zheng Ruan, Junuk Lee, Yangyang Li, Juan Du, Wei Lü.
eLife. (2024) May 23: 13:pr95118. DOI: 10.7554/eLife.95118.3.
Protein phosphorylation is one of the major molecular mechanisms regulating protein activity and function throughout the cell. Pannexin 1 (PANX1) is a large-pore channel permeable to ATP and other cellular metabolites. Its tyrosine phosphorylation and subsequent activation have been found to play critical roles in diverse cellular conditions, including neuronal cell death, acute inflammation, and smooth muscle contraction. Specifically, the non-receptor kinase Src has been reported to phosphorylate Tyr198 and Tyr308 of mouse PANX1 (equivalent to Tyr199 and Tyr309 of human PANX1), resulting in channel opening and ATP release. Although the Src-dependent PANX1 activation mechanism has been widely discussed in the literature, independent validation of the tyrosine phosphorylation of PANX1 has been lacking. Here, we show that commercially available antibodies against the two phosphorylation sites mentioned above—which were used to identify endogenous PANX1 phosphorylation at these two sites—are nonspecific and should not be used to interpret results related to PANX1 phosphorylation. We further provide evidence that neither tyrosine residue is a major phosphorylation site for Src kinase in heterologous expression systems. We call on the field to re-examine the existing paradigm of tyrosine phosphorylation-dependent activation of the PANX1 channel.
Ljubica Mihaljević*, Zheng Ruan*, James Osei-Owusu, Wei Lü, Zhaozhu Qiu.
eLife. (2023) Jan 12: 12:e83935. DOI: 10.7554/eLife.83935.
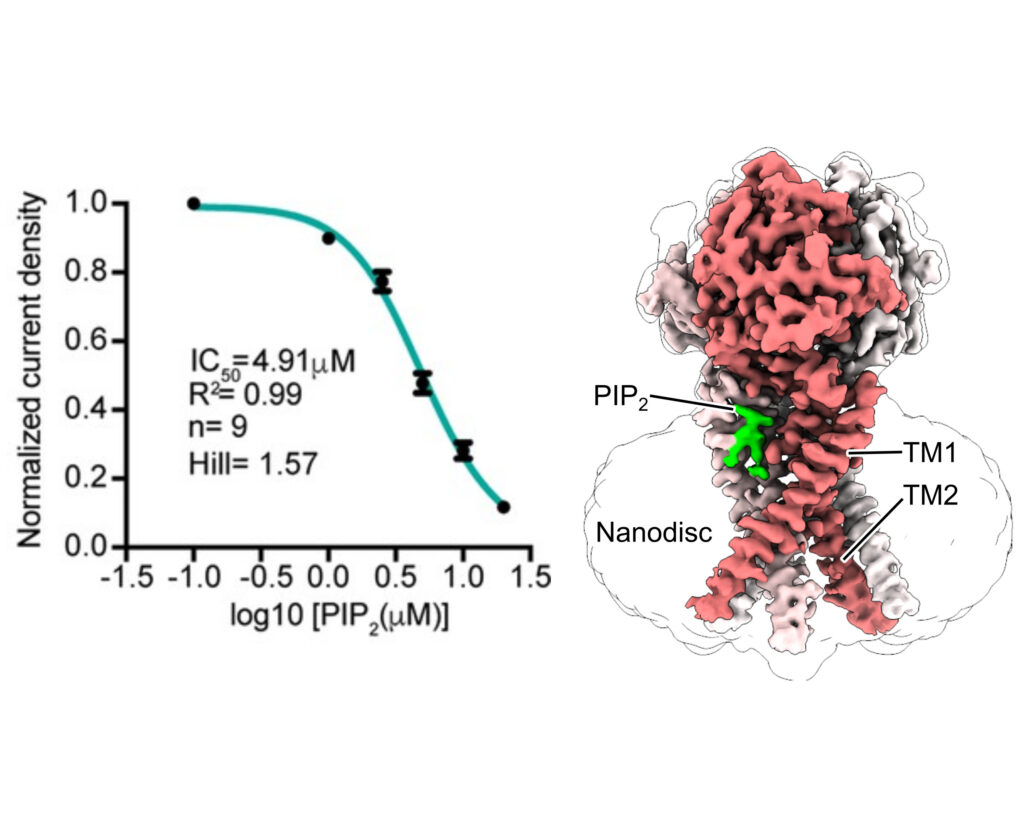
Proton-activated chloride (PAC) channel is a ubiquitously expressed pH-sensing ion channel, encoded by PACC1 (TMEM206). PAC regulates endosomal acidification and macropinosome shrinkage by releasing chloride from the organelle lumens. It is also found at the cell surface, where it is activated under pathological conditions related to acidosis and contributes to acid-induced cell death. However, the pharmacology of the PAC channel is poorly understood. Here, we report that phosphatidylinositol (4,5)-bisphosphate (PIP2) potently inhibits PAC channel activity. We solved the cryo-electron microscopy structure of PAC with PIP2 at pH 4.0 and identified its putative binding site, which, surprisingly, locates on the extracellular side of the transmembrane domain (TMD). While the overall conformation resembles the previously resolved PAC structure in the desensitized state, the TMD undergoes remodeling upon PIP2-binding. Structural and electrophysiological analyses suggest that PIP2 inhibits the PAC channel by stabilizing the channel in a desensitized-like conformation. Our findings identify PIP2 as a new pharmacological tool for the PAC channel and lay the foundation for future drug discovery targeting this channel.
James Osei-Owusu*, Zheng Ruan*, Daniel S. Matasic, Wei Lü, Zhaozhu Qiu.
eLife. (2022) Dec 22; 11:e82955. DOI: 10.7554/eLife.82955.
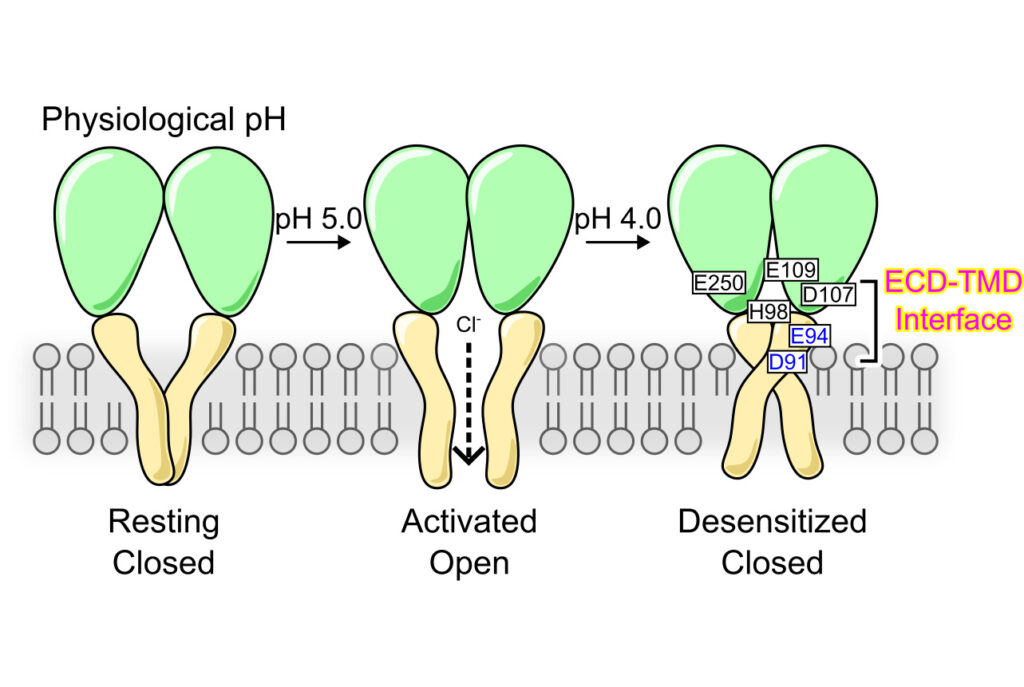
Desensitization is a common property of membrane receptors, including ion channels. The newly identified proton-activated chloride (PAC) channel plays an important role in regulating the pH and size of organelles in the endocytic pathway, and is also involved in acid-induced cell death. However, how the PAC channel desensitizes is largely unknown. Here, we show by patch-clamp electrophysiological studies that PAC (also known as TMEM206/ASOR) undergoes pH-dependent desensitization upon prolonged acid exposure. Through structure-guided and comprehensive mutagenesis, we identified several residues critical for PAC desensitization, including histidine (H) 98, glutamic acid (E) 94, and aspartic acid (D) 91 at the extracellular extension of the transmembrane helix 1 (TM1), as well as E107, D109, and E250 at the extracellular domain (ECD)–transmembrane domain (TMD) interface. Structural analysis and molecular dynamic simulations revealed extensive interactions between residues at the TM1 extension and those at the ECD–TMD interface. These interactions likely facilitate PAC desensitization by stabilizing the desensitized conformation of TM1, which undergoes a characteristic rotational movement from the resting and activated states to the desensitized state. Our studies establish a new paradigm of channel desensitization in this ubiquitously expressed ion channel and pave the way for future investigation of its relevance in cellular physiology and disease.
James Osei-Owusu, Ekaterina Kots, Zheng Ruan, Ljubica Mihaljević, Kevin Hong Chen, Ami Tamhaney, Xinyu Ye, Wei Lu, Harel Weinstein, Zhaozhu Qiu.
PNAS. (2022) Jul 25: 119(31) e2200727119. DOI: 10.1073/pnas.2200727119.
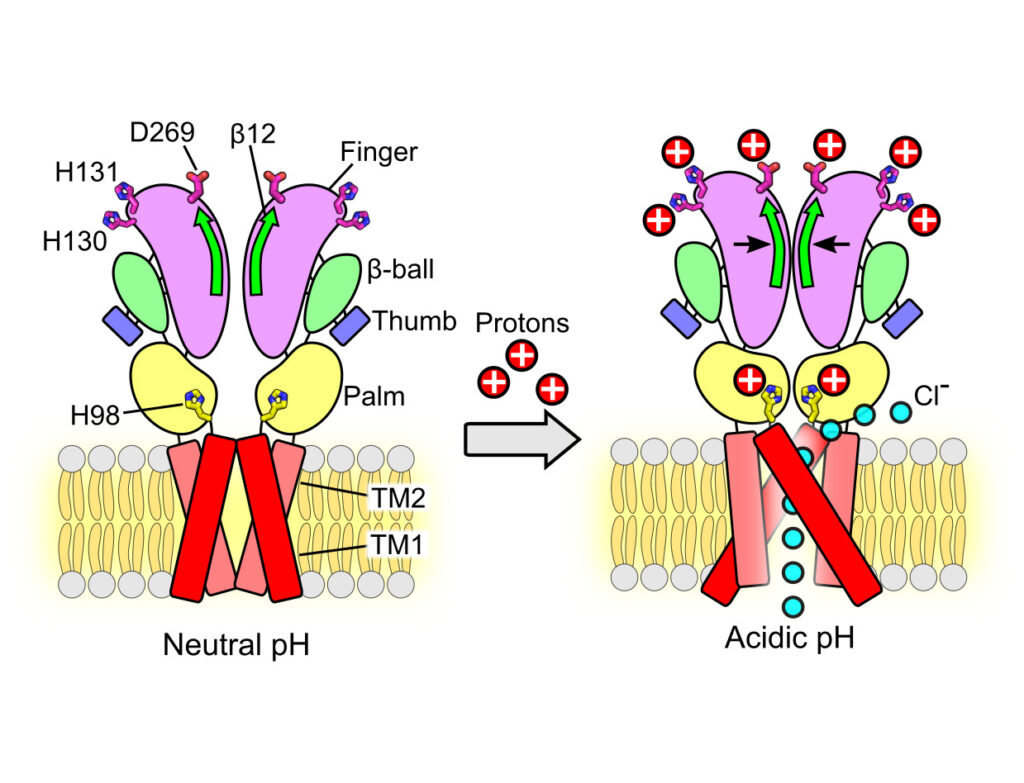
In response to acidic pH, the widely expressed proton-activated chloride (PAC) channel opens and conducts anions across cellular membranes. By doing so, PAC plays an important role in both cellular physiology (endosome acidification) and diseases associated with tissue acidosis (acid-induced cell death). Despite the available structural information, how proton binding in the extracellular domain (ECD) leads to PAC channel opening remains largely unknown. Here, through comprehensive mutagenesis and electrophysiological studies, we identified several critical titratable residues, including two histidine residues (H130 and H131) and an aspartic acid residue (D269) at the distal end of the ECD, together with the previously characterized H98 at the transmembrane domain–ECD interface, as potential pH sensors for human PAC. Mutations of these residues resulted in significant changes in pH sensitivity. Some combined mutants also exhibited large basal PAC channel activities at neutral pH. By combining molecular dynamics simulations with structural and functional analysis, we further found that the β12 strand at the intersubunit interface and the associated “joint region” connecting the upper and lower ECDs allosterically regulate the proton-dependent PAC activation. Our studies suggest a distinct pH-sensing and gating mechanism of this new family of ion channels sensitive to acidic environment.
Zheng Ruan*, Emery Haley*, Ian Orozco*, Rebecca Roth, Mark Sabat, Richard Myers, Wei Lu#, Juan Du#.
Nat Struct Mol Biol. (2021) Jun 24: 28(7):604-613. DOI: 10.1038/s41594-021-00607-4.
The Ca2+-activated TRPM5 channel plays essential roles in taste perception and insulin secretion. However, the mechanism by which Ca2+ regulates TRPM5 activity remains elusive. We report cryo-EM structures of the zebrafish TRPM5 in an apo closed state, a Ca2+-bound open state, and an antagonist-bound inhibited state. We define two novel ligand binding sites: a Ca2+ site (CaICD) in the intracellular domain and an antagonist site in the transmembrane domain (TMD). The CaICD site is unique to TRPM5 and has two roles: modulating the voltage dependence and promoting Ca2+ binding to the CaTMD site, which is conserved throughout TRPM channels. Conformational changes initialized from both Ca2+ sites cooperatively open the ion-conducting pore. The antagonist NDNA wedges into the space between the S1–S4 domain and pore domain, stabilizing the transmembrane domain in an apo-like closed state. Our results lay the foundation for understanding the voltage-dependent TRPM channels and developing new therapeutic agents.
如果你在中国大陆,请点击此链接观看视频。
James Osei-Owusu, Junhua Yang, Ka Ho Leung, Zheng Ruan, Wei Lü, Yamuna Krishnan, Zhaozhu Qiu.
Cell Reports. (2021) Jan 26: 34(4). DOI: 10.1016/j.celrep.2020.108683.
During vesicular acidification, chloride (Cl−), as the counterion, provides the electrical shunt for proton pumping by the vacuolar H+ ATPase. Intracellular CLC transporters mediate Cl− influx to the endolysosomes through their 2Cl−/H+ exchange activity. However, whole-endolysosomal patch-clamp recording also revealed a mysterious conductance releasing Cl− from the lumen. It remains unknown whether CLCs or other Cl− channels are responsible for this activity. Here, we show that the newly identified proton-activated Cl− (PAC) channel traffics from the plasma membrane to endosomes via the classical YxxL motif. PAC deletion abolishes the endosomal Cl− conductance, raises luminal Cl− level, lowers luminal pH, and increases transferrin receptor-mediated endocytosis. PAC overexpression generates a large endosomal Cl− current with properties similar to those of endogenous conductance, hypo-acidifies endosomal pH, and reduces transferrin uptake. We propose that the endosomal Cl− PAC channel functions as a low pH sensor and prevents hyper-acidification by releasing Cl− from the lumen.

Zheng Ruan*, James Osei-Owusu*, Juan Du, Zhaozhu Qiu#, Wei Lü#.
Nature. (2020) Nov 04: 588(7837): 350-354. DOI: 10.1038/s41586-020-2875-7.
The proton-activated chloride channel (PAC) is active across a wide range of mammalian cells and is involved in acid-induced cell death and tissue injury. PAC has recently been shown to represent a novel and evolutionarily conserved protein family. Here we present two cryo-electron microscopy structures of human PAC in a high-pH resting closed state and a low-pH proton-bound non-conducting state. PAC is a trimer in which each subunit consists of a transmembrane domain (TMD), which is formed of two helices (TM1 and TM2), and an extracellular domain (ECD). Upon a decrease of pH from 8 to 4, we observed marked conformational changes in the ECD–TMD interface and the TMD. The rearrangement of the ECD–TMD interface is characterized by the movement of the histidine 98 residue, which is, after acidification, decoupled from the resting position and inserted into an acidic pocket that is about 5 Å away. Within the TMD, TM1 undergoes a rotational movement, switching its interaction partner from its cognate TM2 to the adjacent TM2. The anion selectivity of PAC is determined by the positively charged lysine 319 residue on TM2, and replacing lysine 319 with a glutamate residue converts PAC to a cation-selective channel. Our data provide a glimpse of the molecular assembly of PAC, and a basis for understanding the mechanism of proton-dependent activation.
如果你在中国大陆,请点击此链接观看视频。
Zheng Ruan, Ian Orozco, Juan Du#, Wei Lü#.
Nature. (2020) Jun 03: 584(7822):646-651. DOI: 10.1038/s41586-020-2357-y.
Pannexin 1 (PANX1) is an ATP-permeable channel with critical roles in a variety of physiological functions such as blood pressure regulation, apoptotic cell clearance and human oocyte development. Here we present several structures of human PANX1 in a heptameric assembly at resolutions of up to 2.8 angström, including an apo state, a caspase-7-cleaved state and a carbenoxolone-bound state. We reveal a gating mechanism that involves two ion-conducting pathways. Under normal cellular conditions, the intracellular entry of the wide main pore is physically plugged by the C-terminal tail. Small anions are conducted through narrow tunnels in the intracellular domain. These tunnels connect to the main pore and are gated by a long linker between the N-terminal helix and the first transmembrane helix. During apoptosis, the C-terminal tail is cleaved by caspase, allowing the release of ATP through the main pore. We identified a carbenoxolone-binding site embraced by W74 in the extracellular entrance and a role for carbenoxolone as a channel blocker. We identified a gap-junction-like structure using a glycosylation-deficient mutant, N255A. Our studies provide a solid foundation for understanding the molecular mechanisms underlying the channel gating and inhibition of PANX1 and related large-pore channels.
如果你在中国大陆,请点击此链接观看视频。
Wayland Yeung*, Zheng Ruan*, Natarajan Kannan.
IUBMB Life. (2020) Feb 26: 1-14. DOI: 10.1002/iub.2253.
The faithful propagation of cellular signals in most organisms relies on the coordinated functions of a large family of protein kinases that share a conserved catalytic domain. The catalytic domain is a dynamic scaffold that undergoes large conformational changes upon activation. Most of these conformational changes, such as movement of the regulatory αC-helix from an “out” to “in” conformation, hinge on a conserved, but understudied, loop termed the αC-β4 loop, which mediates conserved interactions to tether flexible structural elements to the kinase core. We previously showed that the αC-β4 loop is a unique feature of eukaryotic protein kinases. Here, we review the emerging roles of this loop in kinase structure, function, regulation, and diseases. Through a kinome-wide analysis, we define the boundaries of the loop for the first time and show that sequence and structural variation in the loop correlate with conformational and regulatory variation. Many recurrent disease mutations map to the αC-β4 loop and contribute to drug resistance and abnormal kinase activation by relieving key auto-inhibitory interactions associated with αC-helix and inter-lobe movement. The αC-β4 loop is a hotspot for post-translational modifications, protein–protein interaction, and Hsp90 mediated folding. Our kinome-wide analysis provides insights for hypothesis-driven characterization of understudied kinases and the development of allosteric protein kinase inhibitors.

Yu-Yang Jiang, Wolfgang Maier, Ralf Baumeister, Gregory Minevich, Ewa Joachimiak, Dorota Wloga, Zheng Ruan, Natarajan Kannan, Stephen Bocarro, Anoosh Bahraini, Krishna Kumar Vasudevan, Karl Lechtreck, Eduardo Orias, Jacek Gaertig.
PLoS Genet. (2019) Jul 24: 15(7). DOI: 10.1371/journal.pgen.1008099.
The length of cilia is controlled by a poorly understood mechanism that involves members of the conserved RCK kinase group, and among them, the LF4/MOK kinases. The multiciliated protist model, Tetrahymena, carries two types of cilia (oral and locomotory) and the length of the locomotory cilia is dependent on their position with the cell. In Tetrahymena, loss of an LF4/MOK ortholog, LF4A, lengthened the locomotory cilia, but also reduced their number. Without LF4A, cilia assembled faster and showed signs of increased intraflagellar transport (IFT). Consistently, overproduced LF4A shortened cilia and downregulated IFT. GFP-tagged LF4A, expressed in the native locus and imaged by total internal reflection microscopy, was enriched at the basal bodies and distributed along the shafts of cilia. Within cilia, most LF4A-GFP particles were immobile and a few either diffused or moved by IFT. We suggest that the distribution of LF4/MOK along the cilium delivers a uniform dose of inhibition to IFT trains that travel from the base to the tip. In a longer cilium, the IFT machinery may experience a higher cumulative dose of inhibition by LF4/MOK. Thus, LF4/MOK activity could be a readout of cilium length that helps to balance the rate of IFT-driven assembly with the rate of disassembly at steady state. We used a forward genetic screen to identify a CDK-related kinase, CDKR1, whose loss-of-function suppressed the shortening of cilia caused by overexpression of LF4A, by reducing its kinase activity. Loss of CDKR1 alone lengthened both the locomotory and oral cilia. CDKR1 resembles other known ciliary CDK-related kinases: LF2 of Chlamydomonas, mammalian CCRK and DYF-18 of C. elegans, in lacking the cyclin-binding motif and acting upstream of RCKs. The new genetic tools we developed here for Tetrahymena have potential for further dissection of the principles of cilia length regulation in multiciliated cells.

Yu-Yang Jiang*, Wolfgang Maier*, Ralf Baumeister, Ewa Joachimiak, Zheng Ruan, Natarajan Kannan, Diamond Clark, Panagiota Louka, Mayukh Guha, Joseph Frankel, Jacek Gaertig.
Genetics. (2019) Feb 1; 211(2) 651-663. DOI: 10.1534/genetics.118.301889.
In a single cell, ciliates maintain a complex pattern of cortical organelles that are arranged along the anteroposterior and circumferential axes. The underlying molecular mechanisms of intracellular pattern formation in ciliates are largely unknown. Ciliates divide by tandem duplication, a process that remodels the parental cell into two daughters aligned head-to-tail. In the elo1-1 mutant of Tetrahymena thermophila, the segmentation boundary/division plane forms too close to the posterior end of the parental cell, producing a large anterior and a small posterior daughter cell, respectively. We show that ELO1 encodes a Lats/NDR kinase that marks the posterior segment of the cell cortex, where the division plane does not form in the wild-type. Elo1 acts independently of CdaI, a Hippo/ Mst kinase that marks the anterior half of the parental cell, and whose loss shifts the division plane anteriorly. We propose that, in Tetrahymena, two antagonistic Hippo circuits focus the segmentation boundary/division plane at the equatorial position, by excluding divisional morphogenesis from the cortical areas that are too close to cell ends.
Sam A. Jamieson*, Zheng Ruan*, Abigail E. Burgess, Jack R. Curry, Hamish D. McMillan, Jodi Brewster, Anita K. Dunbier, Natarajan Kannan, Peter D. Mace.
Sci Signal. 2018 Sep 25; 11(549) DOI: 10.1126/scisignal.aau0597.
The Tribbles family of pseudokinases recruits substrates to the ubiquitin ligase COP1 to facilitate ubiquitylation. CCAAT/enhancer-binding protein (C/EBP) family transcription factors are crucial Tribbles substrates in adipocyte and myeloid cell development. We found that the TRIB1 pseudokinase was able to recruit various C/EBP family members and that the binding of C/EBPβ was attenuated by phosphorylation. To explain the mechanism of C/EBP recruitment, we solved the crystal structure of TRIB1 in complex with C/EBPα, which revealed that TRIB1 underwent a substantial conformational change relative to its substrate-free structure and bound C/EBPα in a pseudosubstrate-like manner. Crystallographic analysis and molecular dynamics and subsequent biochemical assays showed that C/EBP binding triggered allosteric changes that link substrate recruitment to COP1 binding. These findings offer a view of pseudokinase regulation with striking parallels to bona fide kinase regulation—by means of the activation loop and αC helix—and raise the possibility of small molecules targeting either the activation “loop-in” or “loop-out” conformations of Tribbles pseudokinases.

Zheng Ruan, Natarajan Kannan.
PNAS. 2018 Aug 13; 115 (35) E8162-E8171 DOI: doi.org/10.1073/pnas.1803152115.

Liang-Chin Huang, Karen E. Ross, Harold Drabkin, Krzysztof J. Kochut, Zheng Ruan, Peter D’Eustachio, Daniel McSkimming, Cecilia Arighi, Chuming Chen, Darren Natale, Cynthia Smith, Pascale Gaudet, Cathy Wu, and Natarajan Kannan.
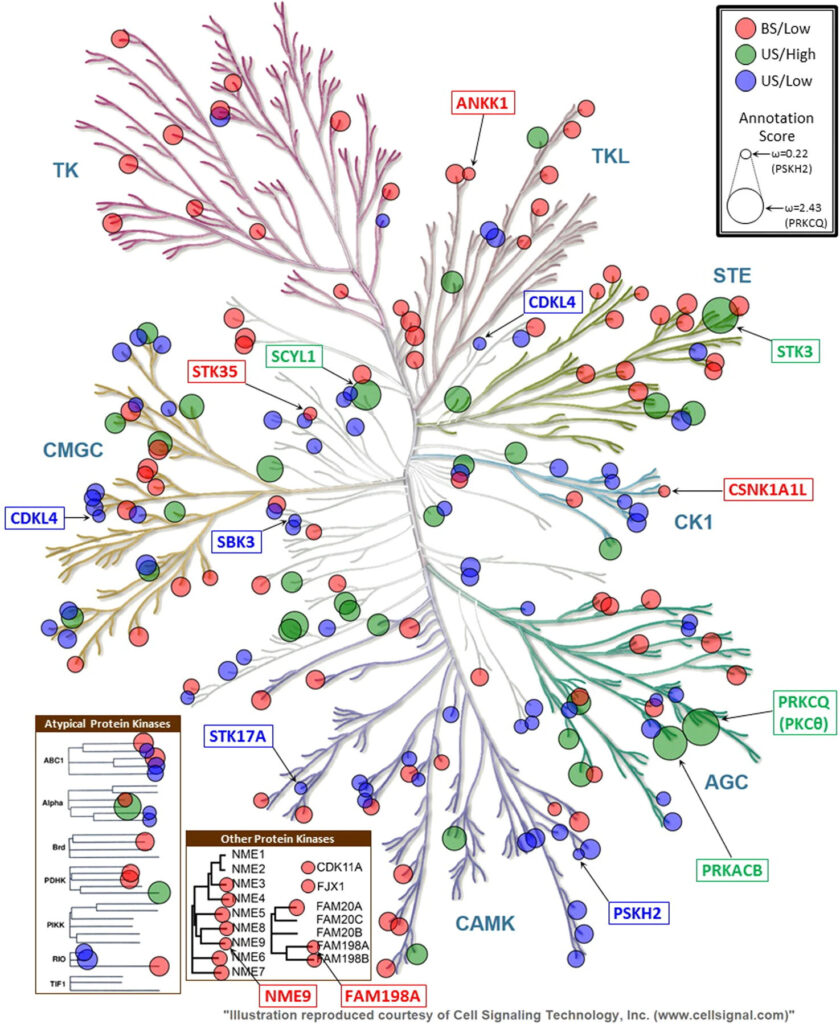
Sci Rep. 2018 Apr 25; 8:6518. DOI: 10.1038/s41598-018-24457-1.
Many bioinformatics resources with unique perspectives on the protein landscape are currently available. However, generating new knowledge from these resources requires interoperable workflows that support cross-resource queries. In this study, we employ federated queries linking information from the Protein Kinase Ontology, iPTMnet, Protein Ontology, neXtProt, and the Mouse Genome Informatics to identify key knowledge gaps in the functional coverage of the human kinome and prioritize understudied kinases, cancer variants and post-translational modifications (PTMs) for functional studies. We identify 32 functional domains enriched in cancer variants and PTMs and generate mechanistic hypotheses on overlapping variant and PTM sites by aggregating information at the residue, protein, pathway and species level from these resources. We experimentally test the hypothesis that S768 phosphorylation in the C-helix of EGFR is inhibitory by showing that oncogenic variants altering S768 phosphorylation increase basal EGFR activity. In contrast, oncogenic variants altering conserved phosphorylation sites in the ‘hydrophobic motif’ of PKCβII (S660F and S660C) are loss-of-function in that they reduce kinase activity and enhance membrane translocation. Our studies provide a framework for integrative, consistent, and reproducible annotation of the cancer kinomes.
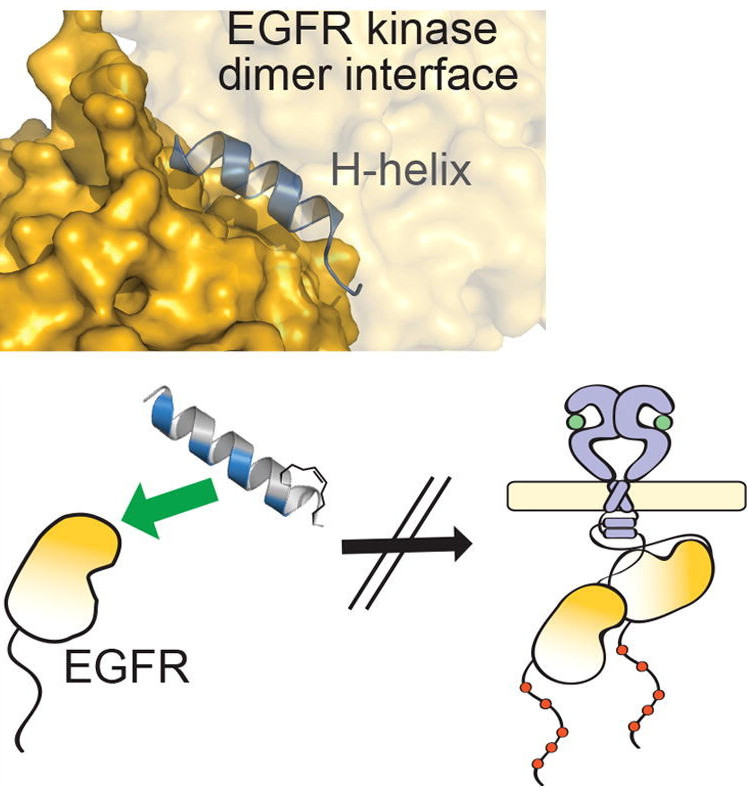
Melody D. Fulton, Laura E. Hanold, Zheng Ruan, Sneha Patel, Aaron M. Beedle, Natarajan Kannan, Eileen J. Kennedy
Bioorg Med Chem. 2018 Mar 15; 26(6):1167-1173. DOI: 10.1016/j.bmc.2017.08.051.
Although EGFR is a highly sought-after drug target, inhibitor resistance remains a challenge. As an alternative strategy for kinase inhibition, we sought to explore whether allosteric activation mechanisms could effectively be disrupted. The kinase domain of EGFR forms an atypical asymmetric dimer via head-to-tail interactions and serves as a requisite for kinase activation. The kinase dimer interface is primarily formed by the H-helix derived from one kinase monomer and the small lobe of the second monomer. We hypothesized that a peptide designed to resemble the binding surface of the H-helix may serve as an effective disruptor of EGFR dimerization and activation. A library of constrained peptides was designed to mimic the H-helix of the kinase domain and interface side chains were optimized using molecular modeling. Peptides were constrained using peptide “stapling” to structurally reinforce an alpha-helical conformation. Peptide stapling was demonstrated to notably enhance cell permeation of an H-helix derived peptide termed EHBI2. Using cell-based assays, EHBI2 was further shown to significantly reduce EGFR activity as measured by EGFR phosphorylation and phosphorylation of the downstream signaling substrate Akt. To our knowledge, this is the first H-helix-based compound targeting the asymmetric interface of the kinase domain that can successfully inhibit EGFR activation and signaling. This study presents a novel, alternative targeting site for allosteric inhibition of EGFR.
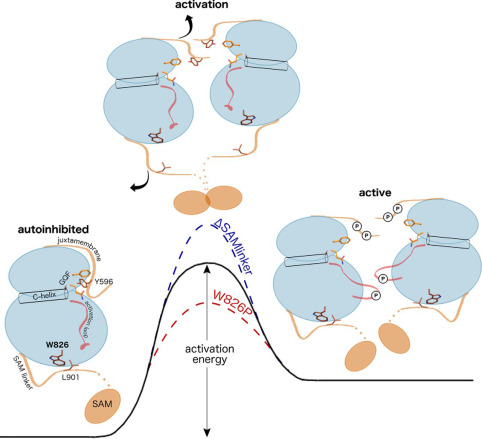
Annie Kwon, Mihir John, Zheng Ruan, Natarajan Kannan.
J Biol Chem. 2018 Apr 6; 293(14): 5102-5116 doi: 10.1074/jbc.RA117.001296.
Ephrin (Eph) receptor tyrosine kinases have evolutionarily diverged from other tyrosine kinases to respond to specific activation and regulatory signals that require close coupling of kinase catalytic and regulatory functions. However, the evolutionary basis for such functional coupling is not fully understood. We employed an evolutionary systems approach involving statistical mining of large sequence and structural data sets to define the hallmarks of Eph kinase evolution and functional specialization. We found that some of the most distinguishing Eph-specific residues structurally tether the flanking juxtamembrane and sterile α motif (SAM) linker regions to the kinase domain, and substitutions of these residues in EphA3 resulted in faster kinase activation. We report for the first time that the SAM domain linker is functionally coupled to the juxtamembrane through co-conserved residues in the kinase domain and that together these residues provide a structural framework for coupling catalytic and regulatory functions. The unique organization of Eph-specific tethering networks and the identification of other Eph-specific sequence features of unknown functions provide new hypotheses for future functional studies and new clues to disease mutations altering Eph kinase–specific functions.
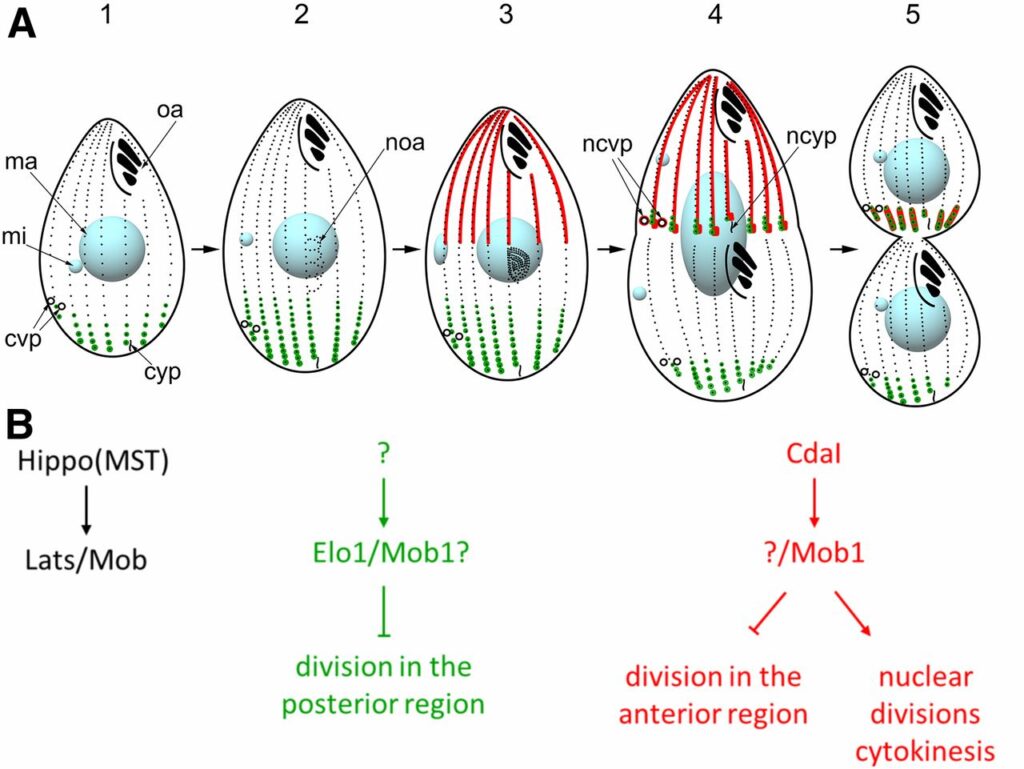
Yu-Yang Jiang, Wolfgang Maier, Ralf Baumeister, Gregory Minevich, Ewa Joachimiak, Zheng Ruan, Natarajan Kannan, Diamond Clarke, Joseph Frankel and Jacek Gaertig.
Genetics. 2017 Jun 1; 206(2):873-888. DOI: doi.org/10.1534/genetics.117.200766.
The mechanisms that govern pattern formation within the cell are poorly understood. Ciliates carry on their surface an elaborate pattern of cortical organelles that are arranged along the anteroposterior and circumferential axes by largely unknown mechanisms. Ciliates divide by tandem duplication: the cortex of the predivision cell is remodeled into two similarly sized and complete daughters. In the conditional cdaI-1 mutant of Tetrahymena thermophila, the division plane migrates from its initially correct equatorial position toward the cell’s anterior, resulting in unequal cell division, and defects in nuclear divisions and cytokinesis. We used comparative whole genome sequencing to identify the cause of cdaI-1 as a mutation in a Hippo/Mst kinase. CdaI is a cortical protein with a cell cycle-dependent, highly polarized localization. Early in cell division, CdaI marks the anterior half of the cell, and later concentrates at the posterior end of the emerging anterior daughter. Despite the strong association of CdaI with the new posterior cell end, the cdaI-1 mutation does not affect the patterning of the new posterior cortical organelles. We conclude that, in Tetrahymena, the Hippo pathway maintains an equatorial position of the fission zone, and, by this activity, specifies the relative dimensions of the anterior and posterior daughter cell.
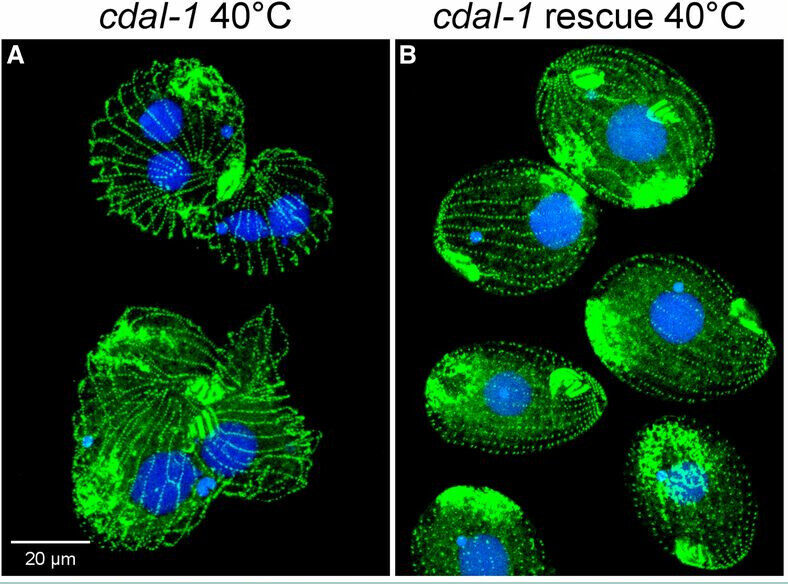
Zheng Ruan, Samiksha Katiyar, Natarajan Kannan.
Biochemistry. 2017 Jan 10; 56(1): 22-32. DOI: 10.1021/acs.biochem.6b00572.
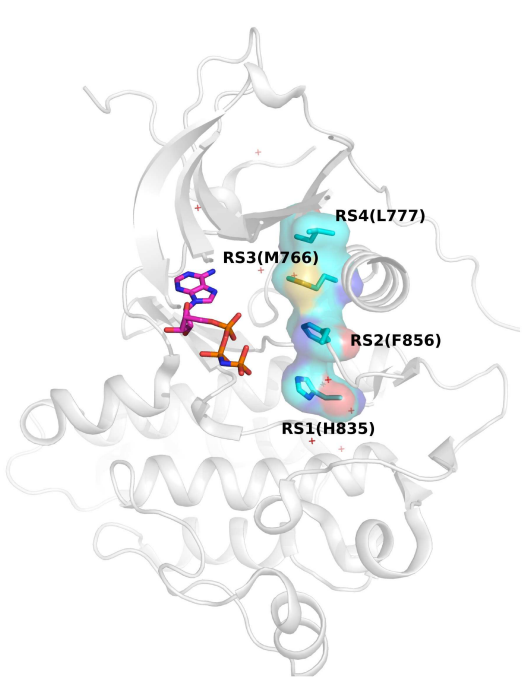
The catalytic activation of protein kinases requires precise positioning of key conserved catalytic and regulatory motifs in the kinase core. The Regulatory Spine (RS) is one such structural motif that is dynamically assembled upon kinase activation. The RS is also a mutational hotspot in cancers; however, the mechanisms by which cancer mutations impact RS assembly and kinase activity are not fully understood. In this study, through mutational analysis of patient derived mutations in the RS of EGFR kinase, we identify an activating mutation, M766T, at the RS3 position. RS3 is located in the regulatory αC-helix, and a series of mutations at the RS3 position suggest a strong correlation between the amino acid type present at the RS3 position and ligand (EGF) independent EGFR activation. Small polar amino acids increase ligand independent activity, while large aromatic amino acids decrease kinase activity. M766T relies on the canonical asymmetric dimer for full activation. Molecular modeling and molecular dynamics simulations of WT and mutant EGFR suggest a model in which M766T activates the kinase domain by disrupting conserved autoinhibitory interactions between M766 and hydrophobic residues in the activation segment. In addition, a water mediated hydrogen bond network between T766, the conserved K745-E762 salt bridge, and the backbone amide of the DFG motif is identified as a key determinant of M766T-mediated activation. M766T is resistant to FDA approved EGFR inhibitors such as gefitinib and erlotinib, and computational estimation of ligand binding free energy identifies key residues associated with drug sensitivity. In sum, our studies suggest an unusual mode of RS assembly and oncogenic EGFR activation, and provide new clues for the design of allosteric protein kinase inhibitors.
Jonathan A Stefely, Floriana Licitra, Leila Laredj, Andrew G Reidenbach, Zachary A Kemmerer, Anais Grangeray, Tiphaine Jaeg-Ehret, Catherine E Minogue, Arne Ulbrich, Paul D Hutchins, Emily M Wilkerson, Zheng Ruan, Deniz Aydin, Alexander S Hebert, Xiao Guo, Elyse C Freiberger, Laurence Reutenauer, Adam Jochem, Maya Chergova, Isabel E Johnson, Danielle C Lohman, Matthew JP Rush, Nicholas W Kwiecien, Pankaj K Singh, Anna I Schlagowski, Brendan J Floyd, Ulrika Forsman, Pavel J Sindelar, Michael S Westphall, Fabien Pierrel, Joffrey Zoll, Matteo Dal Peraro, Natarajan Kannan, Craig A Bingman, Joshua J Coon, Philippe Isope, Hélène Puccio, David J Pagliarini.
Mol Cell. 2016 Aug 18; 63(4): 608-20. DOI: 10.1016/j.molcel.2016.06.030.
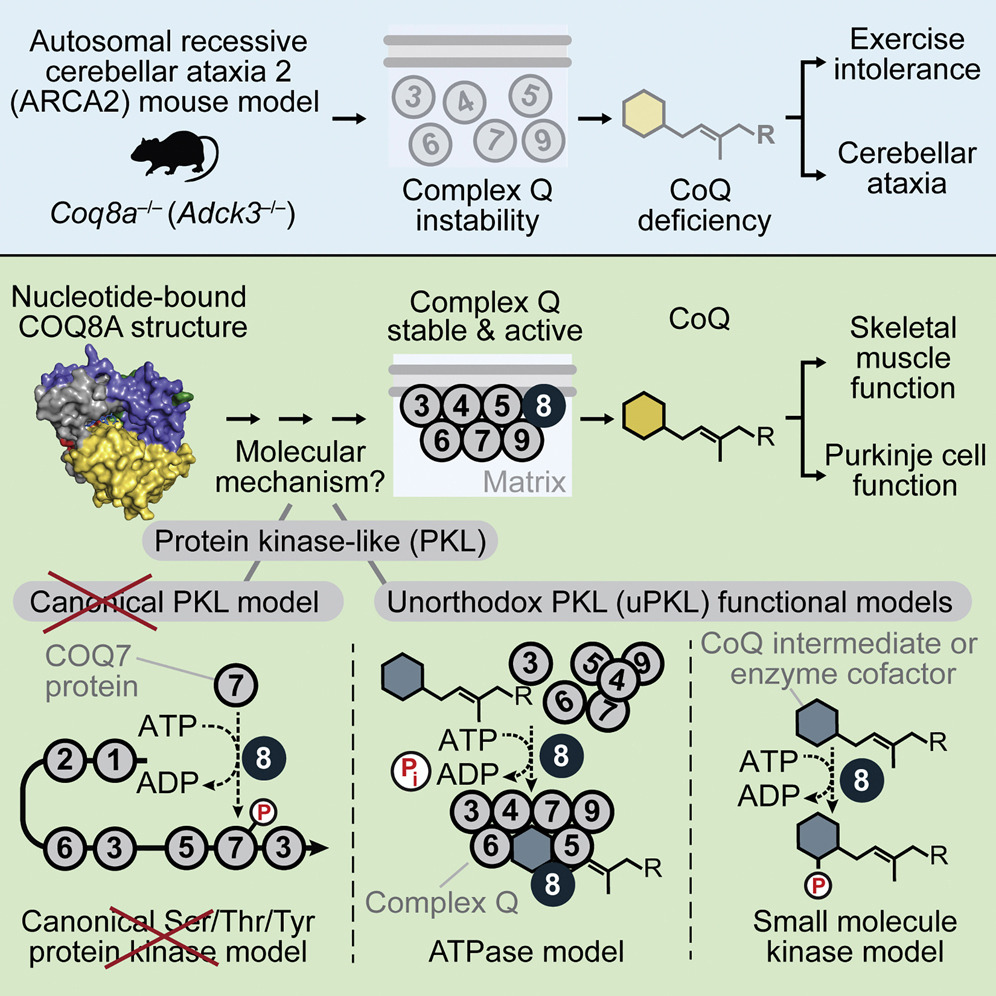
The UbiB protein kinase-like (PKL) family is widespread, comprising one-quarter of microbial PKLs and five human homologs, yet its biochemical activities remain obscure. COQ8A (ADCK3) is a mammalian UbiB protein associated with ubiquinone (CoQ) biosynthesis and an ataxia (ARCA2) through unclear means. We show that mice lacking COQ8A develop a slowly progressive cerebellar ataxia linked to Purkinje cell dysfunction and mild exercise intolerance, recapitulating ARCA2. Interspecies biochemical analyses show that COQ8A and yeast Coq8p specifically stabilize a CoQ biosynthesis complex through unorthodox PKL functions. Although COQ8 was predicted to be a protein kinase, we demonstrate that it lacks canonical protein kinase activity in trans. Instead, COQ8 has ATPase activity and interacts with lipid CoQ intermediates, functions that are likely conserved across all domains of life. Collectively, our results lend insight into the molecular activities of the ancient UbiB family and elucidate the biochemical underpinnings of a human disease.
Smita Mohanty, Krishnadev Oruganty, Annie Kwon, Dominic P Byrne, Samantha Ferries, Zheng Ruan, Laura E Hanold, Samiksha Katiyar, Eileen J Kennedy, Patrick A Eyers, Natarajan Kannan.
PLoS Genet. 2016 Feb 29; 12(2): e1005885. DOI: 10.1371/journal.pgen.1005885.
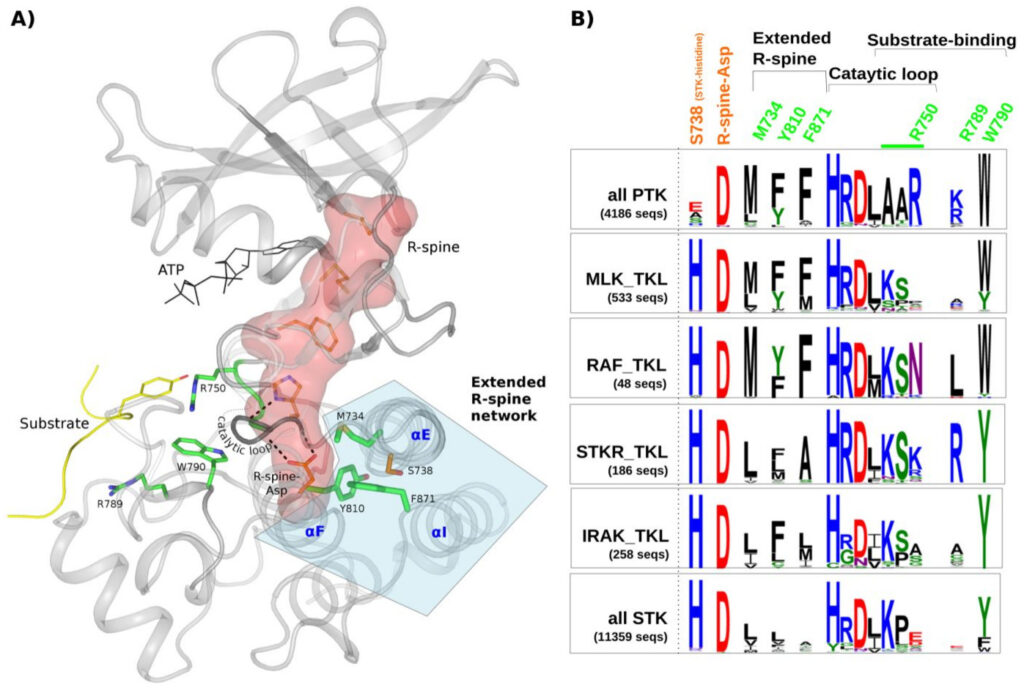
Protein tyrosine kinases (PTKs) are a group of closely related enzymes that have evolutionarily diverged from serine/threonine kinases (STKs) to regulate pathways associated with multi-cellularity. Evolutionary divergence of PTKs from STKs has occurred through accumulation of mutations in the active site as well as in the commonly conserved hydrophobic core. While the functional significance of active site variations is well understood, relatively little is known about how hydrophobic core variations contribute to PTK evolutionary divergence. Here, using a combination of statistical sequence comparisons, molecular dynamics simulations, mutational analysis and in vitro thermostability and kinase assays, we investigate the structural and functional significance of key PTK-specific variations in the kinase core. We find that the nature of residues and interactions in the hydrophobic core of PTKs is strikingly different from other protein kinases, and PTK-specific variations in the core contribute to functional divergence by altering the stability and dynamics of the kinase domain. In particular, a functionally critical STK-conserved histidine that stabilizes the regulatory spine in STKs is selectively mutated to an alanine, serine or glutamate in PTKs, and this loss-of-function mutation is accommodated, in part, through compensatory PTK-specific interactions in the core. In particular, a PTK-conserved phenylalanine in the I-helix appears to structurally and functionally compensate for the loss of STK-histidine by interacting with the regulatory spine, which has far-reaching effects on enzyme activity, inhibitor sensing, and stability. We propose that hydrophobic core variations provide a selective advantage during PTK evolution by increasing the conformational flexibility, and therefore the allosteric potential of the kinase domain. Our studies also suggest that Tyrosine Kinase Like kinases such as RAF are intermediates in PTK evolutionary divergence inasmuch as they share features of both PTKs and STKs in the core. Finally, our studies provide an evolutionary framework for identifying and characterizing disease and drug resistance mutations in the kinase core.
Zheng Ruan, Natarajan Kannan.
Biochemistry. 2015 Jul 14; 54(27): 4216-25. DOI: 10.1021/acs.biochem.5b00444.
The epidermal growth factor receptor (EGFR) kinase is activated by a variety of mutations in human cancers. R776H is one such recurrent mutation (R752H in another numbering system) in the αC-β4 loop of the tyrosine kinase domain that activates EGFR in the absence of the activating EGF ligand. However, the mechanistic details of how R776H contributes to kinase activation are not well understood. Here using cell-based cotransfection assays, we show that the R776H mutation activates EGFR in a dimerization-dependent manner by preferentially adopting the acceptor position in the asymmetric dimer. The acceptor function, but not the donor function, is enhanced for the R776H mutant, supporting the “superacceptor” hypothesis proposed for oncogenic mutations in EGFR. We also find that phosphorylation of monomeric EGFR is increased by R776H mutation, providing insights into EGFR lateral phosphorylation and oligomerization. On the basis of molecular modeling and molecular dynamics simulation, we propose a model in which loss of key autoinhibitory αC-helix capping interaction and alteration of coconserved cis regulatory interactions between the kinase domain and the flanking regulatory segments contribute to mutational activation. Since the R776 equivalent position is mutated in ErbB2 and ErbB4, our studies have implications for understanding kinase mutational activation in other ErbB family members as well.

Tuan Nguyen, Zheng Ruan, Krishnadev Oruganty, Natarajan Kannan.
PLoS One. 2015; 10(3): e0119636. DOI: 10.1371/journal.pone.0119636.
Mitogen activated protein kinases (MAPKs) form a closely related family of kinases that control critical pathways associated with cell growth and survival. Although MAPKs have been extensively characterized at the biochemical, cellular, and structural level, an integrated evolutionary understanding of how MAPKs differ from other closely related protein kinases is currently lacking. Here, we perform statistical sequence comparisons of MAPKs and related protein kinases to identify sequence and structural features associated with MAPK functional divergence. We show, for the first time, that virtually all MAPK-distinguishing sequence features, including an unappreciated short insert segment in the β4-β5 loop, physically couple distal functional sites in the kinase domain to the D-domain peptide docking groove via the C-terminal flanking tail (C-tail). The coupling mediated by MAPK-specific residues confers an allosteric regulatory mechanism unique to MAPKs. In particular, the regulatory αC-helix conformation is controlled by a MAPK-conserved salt bridge interaction between an arginine in the αC-helix and an acidic residue in the C-tail. The salt-bridge interaction is modulated in unique ways in individual sub-families to achieve regulatory specificity. Our study is consistent with a model in which the C-tail co-evolved with the D-domain docking site to allosterically control MAPK activity. Our study provides testable mechanistic hypotheses for biochemical characterization of MAPK-conserved residues and new avenues for the design of allosteric MAPK inhibitors.

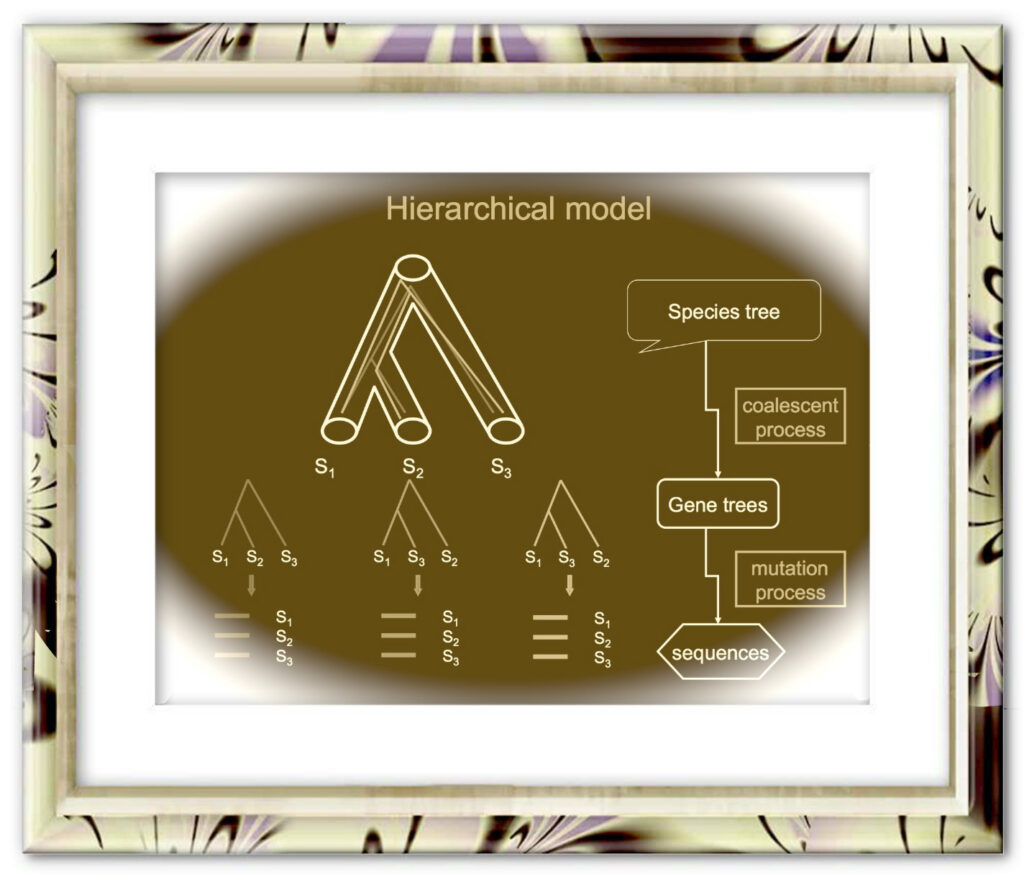
Timothy I Shaw, Zheng Ruan, Travis C Glenn, Liang Liu.
Nucleic Acids Res. 2013 Jul; 41(Web Server issue): W238-W241. DOI: 10.1093/nar/gkt377.
The coalescent methods for species tree reconstruction are increasingly popular because they can accommodate coalescence and multilocus data sets. Herein, we present STRAW, a web server that offers workflows for reconstruction of phylogenies of species using three species tree methods—MP-EST, STAR and NJst. The input data are a collection of rooted gene trees (for STAR and MP-EST methods) or unrooted gene trees (for NJst). The output includes the estimated species tree, modified Robinson-Foulds distances between gene trees and the estimated species tree and visualization of trees to compare gene trees with the estimated species tree. The web sever is available at https://straw.phylolab.net/SpeciesTreeAnalysis/index.php.